<style> .tg {border-collapse:collapse;border-spacing:0;} .tg td{border-color:black;border-style:solid;border-width:1px;font-family:Arial, sans-serif;font-size:18px; overflow:hidden;padding:10px 5px;word-break:normal;} .tg th{border-color:black;border-style:solid;border-width:1px;font-family:Arial, sans-serif;font-size:14px; font-weight:normal;overflow:hidden;padding:10px 5px;word-break:normal;} </style> ### Genomic data standards <a href="https://xkcd.com/2116/"><img src="https://imgs.xkcd.com/comics/norm_normal_file_format.png"> --- ## What is a standard? > A norm or specification applied to a repeated process. It includes a formal definition of terms and a description of formats and expected behavior of a system <p class="fragment">Standards → Interoperability</p> --- What happens when there is no standard (or, equivalently, too many standards) <img src="images/genomic-data-standards/geektech-charging-leads.png" width="600" alt="Geektech charging leads"> --- <img src="https://imgs.xkcd.com/comics/standards.png" width="600"> <img src="images/genomic-data-standards/eu-common-charger.png" width="600" alt="Geektech charging leads" class="fragment"> --- ### Existing standards in genomics - Project metadata - Ontologies - File formats - Reference genomes - Web standards and APIs <p>Standards → Interoperability</p> --- ### GA4GH > The Global Alliance for Genomics and Health (GA4GH) is a policy-framing and technical standards-setting organization, seeking to enable responsible genomic data sharing within a human rights framework. - [ga4gh.org](https://www.ga4gh.org/) - formed in 2013 --- <h3>Research is organized in <i>projects</i></h3> <div class="row"> <div class="col"> <img src="images/genomic-data-standards/tubes.svg" width="450"> </div> <div class="col fragment"> <img src="images/genomic-data-standards/tubes_color.svg" width="450"> </div> </div> <div class="fragment"> How do we conceptualize a research project? </div> --- <h2>Each project has 3 components</h2> <img src="images/genomic-data-standards/data-code-compute.svg" width="500" style="align:center"><br/> --- <h2>Organizing multiple projects is a challenge</h2> <img src="images/genomic-data-standards/linking-projects.svg" width="600" style="align:center"><br/> --- <h2>How do I re-use a component?</h2> <img src="images/genomic-data-standards/linking-projects-reuse.svg" width="600" style="align:center"><br/> --- <h2>A project is a set of edges in a tripartite graph</h2> <img src="images/genomic-data-standards/linking-projects-modular.svg" width="600" style="align:center"><br/><br/> --- <h2>Enable linking with interfaces</h2> <img src="images/genomic-data-standards/linking-interfaces.svg" width="600" style="align:center"><br/> --- ### Existing standards in genomics - Project metadata (Data → Code) - Ontologies (Data → Code) - File formats (Data → Data) - Reference genomes (Data → Data) - Web standards and APIs (Data → Compute) <p>Standards → Interoperability</p> --- <h2>PEP: Portable Encapsulated Projects</h2> <img src="images/genomic-data-standards/pep_center_white.svg" width="700"> --- <img src="images/genomic-data-standards/pep_contents_white.svg" width="750"> --- <div class="bullet"> <h2><img src="images/genomic-data-standards/pep_logo.svg" width="70">PEP format</h2> </div> <div class="bullet"> <img src="images/genomic-data-standards/file.svg" width="30">project_config.yaml </div> <pre><code>sample_table: /path/to/samples.tsv output_dir: /path/to/output/folder </code></pre> <hr> <div class="bullet"> <img src="images/genomic-data-standards/file.svg" width="30">samples.csv </div> <pre><code>sample_name, protocol, organism, data_source frog_0h, RNA-seq, frog, /path/to/frog0.gz frog_1h, RNA-seq, frog, /path/to/frog1.gz frog_2h, RNA-seq, frog, /path/to/frog2.gz frog_3h, RNA-seq, frog, /path/to/frog3.gz </code></pre> --- <h3>PEP portability features</h3> <div style="text-align: left"> <!-- <span class="bullet"><img src="images/genomic-data-standards/ftnetwork-connected.svg" width="50" class="bullet">Project API</span><br> --> <span class="bullet"><img src="images/genomic-data-standards/replace_white.svg" width="50" class="bullet">Derived attributes</span><br> <span class="bullet"><img src="images/genomic-data-standards/implies_white.svg" width="50" class="bullet">Implied attributes</span><br> <span class="bullet"><img src="images/genomic-data-standards/subproject_white.svg" width="50" class="bullet">Subprojects</span><br> </div> --- <span class="bullet"><img src="images/genomic-data-standards/replace_white.svg" width="50" class="bullet">Derived attributes</span><br> <div class="well">Build new sample attributes from existing ones</div> <!-- These code data-markdown sections seem to kill data-backgrounds --> Without derived attribute: <code> <div data-markdown style="font-size:0.4em"> | sample_name | t | protocol | organism | data_source | | ------------- | ---- | :-------------: | -------- | ---------------------- | | frog_0h | 0 | RNA-seq | frog | /path/to/frog0.gz | | frog_1h | 1 | RNA-seq | frog | /path/to/frog1.gz | | frog_2h | 2 | RNA-seq | frog | /path/to/frog2.gz | | frog_3h | 3 | RNA-seq | frog | /path/to/frog3.gz | </div> </code> Using derived attribute: <code> <div data-markdown style="font-size:0.4em"> | sample_name | t | protocol | organism | data_source | | ------------- | ---- | :-------------: | -------- | ---------------------- | | frog_0h | 0 | RNA-seq | frog | my_samples | | frog_1h | 1 | RNA-seq | frog | my_samples | | frog_2h | 2 | RNA-seq | frog | my_samples | | frog_3h | 3 | RNA-seq | frog | my_samples | | crab_0h | 0 | RNA-seq | crab | your_samples | | crab_3h | 3 | RNA-seq | crab | your_samples | </div> </code> --- <code> <div data-markdown style="font-size:0.5em"> | sample_name | t | protocol | organism | data_source | | ------------- | ---- | :-------------: | -------- | ---------------------- | | frog_0h | 0 | RNA-seq | frog | my_samples | | frog_1h | 1 | RNA-seq | frog | my_samples | | frog_2h | 2 | RNA-seq | frog | my_samples | | frog_3h | 3 | RNA-seq | frog | my_samples | | crab_0h | 0 | RNA-seq | crab | your_samples | | crab_3h | 3 | RNA-seq | crab | your_samples | </div> </code> Project config file: ```yaml sample_modifiers: derive: attributes: [data_source] sources: my_samples: "/path/to/my/samples/{organism}_{t}h.gz" your_samples: "/your/samples/{organism}_{t}h.gz" ``` {variable} identifies sample annotation columns <div class="well">Benefit: Enables distributed files, portability</div> --- Implied attributes <div class="well">Add new sample attributes conditioned on values of existing attributes</div> <div class="col2"> Before:<br> <code> <div data-markdown style="font-size:0.5em"> | sample_name | protocol | organism | | ------------- | :-------------: | -------- | | human_1 | RNA-seq | human | | human_2 | RNA-seq | human | | human_3 | RNA-seq | human | | mouse_1 | RNA-seq | mouse | </div> </code> </div> <div class="col2"> After:<br> <code> <div data-markdown style="font-size:0.5em"> | sample_name | protocol | organism | genome | | ------------- | :-------------: | -------- | ------ | | human_1 | RNA-seq | human | hg38 | | human_2 | RNA-seq | human | hg38 | | human_3 | RNA-seq | human | hg38 | | mouse_1 | RNA-seq | mouse | mm10 | </div> </code> </div> --- <code> <div data-markdown style="font-size:0.5em"> | sample_name | protocol | organism | | ------------- | :-------------: | -------- | | human_1 | RNA-seq | human | | human_2 | RNA-seq | human | | human_3 | RNA-seq | human | | mouse_1 | RNA-seq | mouse | </div> </code> Project config file: ```yaml sample_modifiers: imply: - if: organism: human then: genome: hg38 - if: organism: mouse then: genome: mm10 ``` <div class="well">Benefit: Divides project from sample metadata</div> --- Project amendments <div class="well">Define activatable project attributes.</div> ```yaml project_modifiers: amend: diverse: sample_table: psa_rrbs_diverse.csv cancer: sample_table: psa_rrbs_intracancer.csv ``` <div class="well">Benefit: Defines multiple similar projects in a single file</div> --- ### Is it enough? Study 1: <code> <div data-markdown style="font-size:0.4em"> | sample_name | t | protocol | organism | data_source | | ------------- | ---- | :-------------: | -------- | ---------------------- | | frog_2h | 2 | RNA-seq | frog | my_samples | | frog_3h | 3 | RNA-seq | frog | my_samples </div> </code> Study 2: <code> <div data-markdown style="font-size:0.4em"> | sample_name | t | protocol | organism | data_source | | ------------- | ---- | :-------------: | -------- | ---------------------- | | frog_0m | 0 | RNAseq | frog | my_samples | | frog_60m | 60 | RNAseq | frog | my_samples | </div> </code> --- ### Ontologies > A formal vocabulary and definition of concepts, entities, and their relationships. 1. Terms 2. Relations --- ## Gene Ontology - 3 Ontologies: - Molecular Function - Cellular Component - Biological Process 1. Terms: [Hexose Biosynthetic Process](http://amigo.geneontology.org/amigo/term/GO:0019319) 2. Relations: is-a, is-part-of, regulates ([Relation Ontology](https://doi.org/10.1186/gb-2005-6-5-r46)) --- 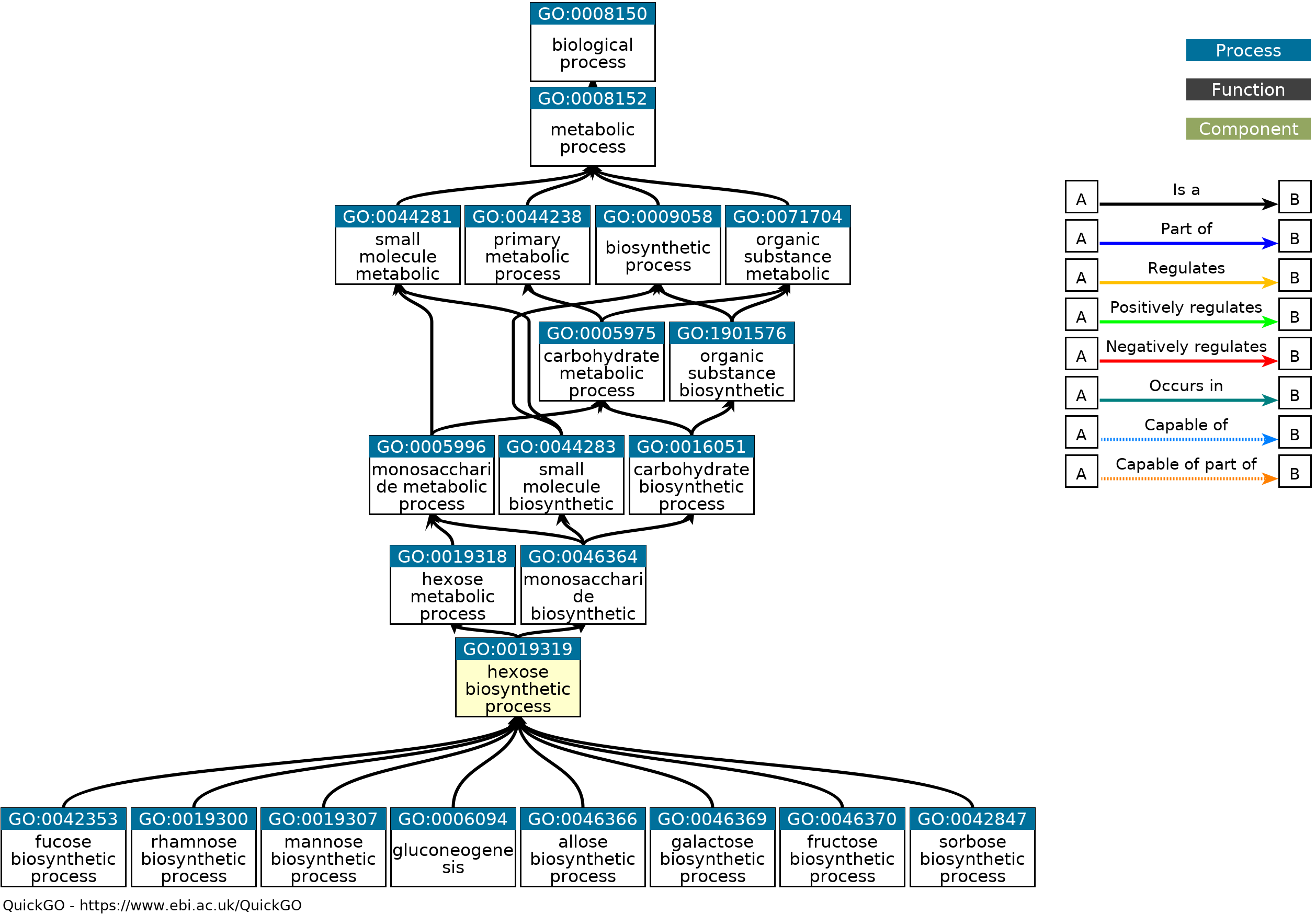 --- ### EDAM Ontology 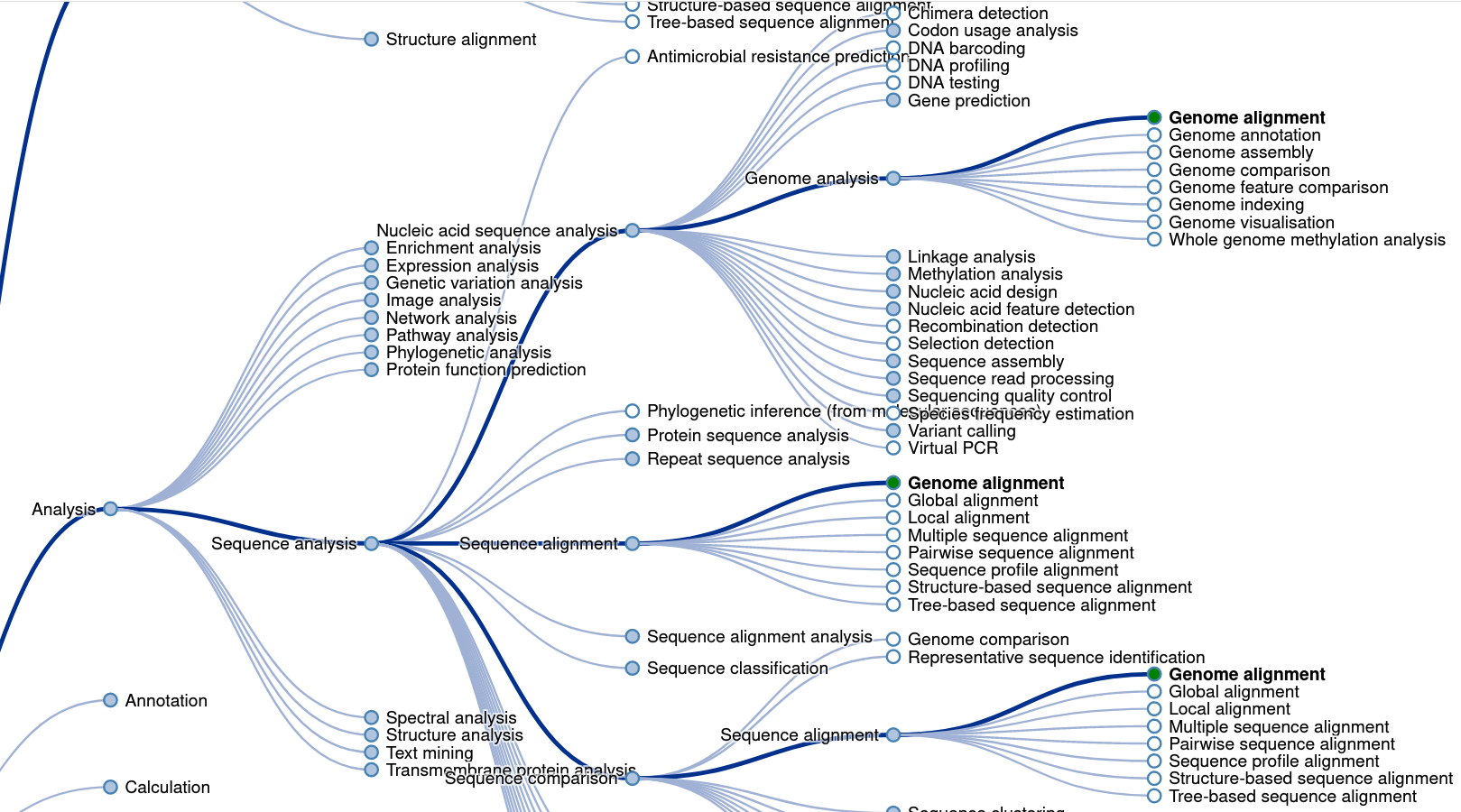 --- ### Sequence Ontology Term: `open_chromatin_region` SO Accession: `SO:0001747` <div class="row"> <div class="col"> 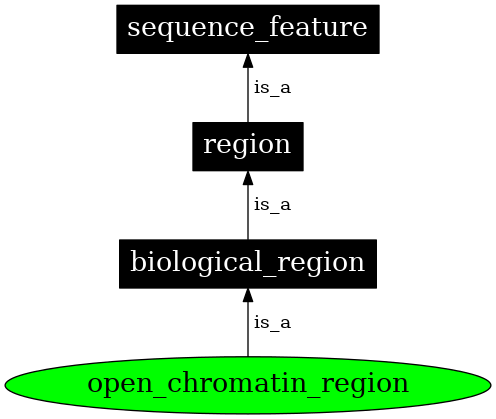 </div> <div class="col"> Definition: A DNA sequence that in the normal state of the chromosome corresponds to an unfolded, un-complexed stretch of double-stranded DNA. </div> </div> --- <img src="images/genomic-data-standards/SO0000162_lg.png" height="670"> --- How can we enforce or validate terms? Study 1: <code> <div data-markdown style="font-size:0.4em"> | sample_name | t | protocol | organism | data_source | | ------------- | ---- | :-------------: | -------- | ---------------------- | | frog_2h | 2 | RNA-seq | frog | my_samples | | frog_3h | 3 | RNA-seq | frog | my_samples </div> </code> Study 2: <code> <div data-markdown style="font-size:0.4em"> | sample_name | t | protocol | organism | data_source | | ------------- | ---- | :-------------: | -------- | ---------------------- | | frog_0m | 0 | RNAseq | frog | my_samples | | frog_60m | 60 | RNAseq | frog | my_samples | </div> </code> --- ### JSON Schema ``` { "productId": 1, "productName": "A green door", "price": 12.50, "tags": [ "home", "green" ] } ``` - What is productId? - Is productName required? - Can the price be zero (0)? - Are all of the tags string values? --- ### RNA-seq analysis ``` { "sample_name": frog_2h, "t": "2", "protocol": "RNA-seq", "organism": "frog", "data_source": "my_samples" } ``` - Is sample_name required? - What is t? - What are the allowable protocols? - What are the allowable organisms? - What is the input file type expected for data source? --- ### Schema ``` { "title": "Product", "description": "A product from Acme's catalog", "type": "object", "properties": { "productId": { "description": "The unique identifier for a product", "type": "integer" }, "productName": { "description": "Name of the product", "type": "string" } }, "required": [ "productId", "productName" ] } ``` --- ### Validating objects with schemas ```shell jsonschema --instance sample.json sample.schema ``` ```python validate([2, 3, 4], {"maxItems": 2}) Traceback (most recent call last): ... ValidationError: [2, 3, 4] is too long ``` --- ### What is a schema? A formal definition of the properties and allowable values for data <span class="fragment"> JSON-Schema provides a standard format for how to describe generic data objects, and tools to validate them. </span> --- ### A schema for ATAC-seq pipeline ``` description: A PEP for ATAC-seq samples for the PEPATAC pipeline. imports: - http://schema.databio.org/pep/2.0.0.yaml properties: samples: type: array items: type: object properties: sample_name: type: string description: "Name of the sample" organism: type: string description: "Organism" protocol: type: string description: "Must be an ATAC-seq or DNAse-seq sample" genome: type: string description: "Refgenie genome registry identifier" read_type: type: string description: "Is this single or paired-end data?" enum: ["SINGLE", "PAIRED"] read1: anyOf: - type: string description: "Fastq file for read 1" - type: array items: type: string read2: anyOf: - type: string description: "Fastq file for read 2 (for paired-end experiments)" - type: array items: type: string required_files: - read1 files: - read1 - read2 required: - sample_name - protocol - read1 - genome required: - samples ``` --- <img src="images/genomic-data-standards/linking-interfaces.svg" width="600" style="align:center"><br/> --- ### Standards: File formats - FASTA: DNA sequences - FASTQ: Short DNA sequencer reads - SAM/BAM: Aligned DNA sequences - BED: genomic intervals - VCF: genomic variation --- ### File formats are not enough Say I have two BED files. Are they comparable? File 1: ``` chr1 15300 17300 chr1 24900 25600 chr1 72420 74440 ``` File 2: ``` 1 18110 19300 1 64000 65600 1 72400 74700 ``` <div class="fragment">sequence names must match</div> <div style="color:black">.</div> --- ### File formats are not enough Say I have two BED files. Are they comparable? File 1: ``` chr1 15300 17300 chr1 24900 25600 chr1 72420 74440 ``` File 2: ``` chr1 18110 19300 chr1 64000 65600 chr1 72400 74700 ``` sequence names must match. How about now? <div class="fragment">reference sequence must be compatible</div> --- <div class="row" style="border: 1px solid white"> <div class="col" style="border: 1px solid white"></div> <div class="col" style="border: 1px solid white">Names Match</div> <div class="col" style="border: 1px solid white">Names Mismatch</div> </div> <div class="row" style="border: 1px solid white"> <div class="col" style="border: 1px solid white">Reference Match</div> <div class="col" style="border: 1px solid white">Great!</div> <div class="col" style="border: 1px solid white">Adjust names?</div> </div> <div class="row" style="border: 1px solid white"> <div class="col" style="border: 1px solid white">Ref. Mismatch</div> <div class="col" style="border: 1px solid white">Don't be fooled!</div> <div class="col" style="border: 1px solid white">No compatibility</div> </div> --- ### Standards: Reference genomes Reference genomes are versioned. human reference: hg18, hg19, hg38/GRCh38, T2T mouse reference: mm8, mm9, mm10 They are not compatible! --- ### Converting among genome builds Liftover ([Luu et al. 2020](https://doi.org/10.1093/nargab/lqaa054)) <img src="images/genomic-data-standards/liftover.jpeg" height="500"> --- ### Standards: Reference genomes There are also many variations of the *same* version. - 3 GRCh38 providers: NCBI, Ensembl, UCSC - hard- soft-, or no repeat masks? - what are the chromosomes named (1 vs chr1 vs chr1, NC_000001.11) - what secondary scaffolds are included? - how is the assembly named (hg38, GRCh38, or GCF_000001405.39)? - are any decoy sequences included (like EBV)? <div class="fragment"> Downstream results are not compatible if based on a different reference! </div> --- Andy Yates' "Genome provider analysis" <table class="tg" style="margin-bottom:60px"> <thead> <tr> <th class="tg">Provider</th> <th class="tg">Chr1 name</th> <th class="tg">Chr1 length</th> <th class="tg">Chr1 md5</th> <th class="tg">Num chroms</th> </tr> </thead> <tbody> <tr> <td class="tg">Ensembl primary</td> <td class="tg">1</td> <td class="tg">248956422</td> <td class="tg">2648ae1bacce4ec4b6cf337dcae37816</td> <td class="tg">195</td> </tr> <tr> <td class="tg">Ensembl toplevel</td> <td class="tg">1</td> <td class="tg">248956422</td> <td class="tg">2648ae1bacce4ec4b6cf337dcae37816</td> <td class="tg">649</td> </tr> <tr> <td class="tg">NCBI</td> <td class="tg">NC_000001.11</td> <td class="tg">248956422</td> <td class="tg">6aef897c3d6ff0c78aff06ac189178dd</td> <td class="tg">640</td> </tr> <tr> <td class="tg">UCSC</td> <td class="tg">chr1</td> <td class="tg">248956422</td> <td class="tg">2648ae1bacce4ec4b6cf337dcae37816</td> <td class="tg">456</td> </tr> </tbody> </table> <div class="small">https://gist.github.com/andrewyatz/692f81baab1bebaf09c481937f2ad6c6</div> --- ### Hashing > A hash function maps an object to a finite space - must be deterministic - should be very fast to compute - should reduce collisions <div class="fragment">Used for hash tables, but also...</div> --- ### Hashing The output, called a *hash* or *digest*, can be seen as a fingerprint or barcode. It is a unique identifier of an input item. <div class="fragment">As long as there are no collisions...</div> --- ### How [GA4GH:refget](https://doi.org/10.1093/bioinformatics/btab524) works <div class="col2"> <img src="images/genomic-data-standards/refget_concept.svg" style="background:white" width="400"> </div> --- ### Sanitize sequence before digesting 1. Remove whitespace 2. Make upper-case --- ### CRAM format Can use refget server for references --- Refget is great, but: - what about *collections* of sequences, like a reference genome? - what about *names* of sequences? --- <h2>Sequence collections</h2> Digest algorithm <img src="images/genomic-data-standards/digest_algorithm.svg" style="background:white" width="900"> --- <h2>Sequence collections</h2> Digest lookup <img src="images/genomic-data-standards/digest_lookup.svg" style="background:white" width="900"> --- hg38 → a6748aa0f --- ✓ File formats ✓ Reference genome Are these RNA-seq experiments comparable? File 1: ``` sample1 sample2 sample3 MIR1302 7 2 153 FAM138A 64 30 55 LINC01409 14 22 17 PRDM16-DT 2 0 0 ``` File 2: ``` sample1 sample2 sample3 ENSG00000243485 64 22 55 ENSG00000237613.2 17 53 52 ENSG00000238009.6 12 93 455 ENST00000477740.5 2 35 7 ``` --- ### Existing standards in genomics - Gene names - HUGO Gene Nomenclature Committee (https://www.genenames.org/) - Gene and transcript annotation - GENCODE (https://www.gencodegenes.org/) --- ### RNA-seq <a href="https://en.wikipedia.org/wiki/RNA-Seq"><img src="https://upload.wikimedia.org/wikipedia/commons/f/f3/Summary_of_RNA-Seq.svg" height="600"></a> --- ### Transcript models  --- <a href="https://doi.org/10.1371/journal.pcbi.1007664 "><img src="images/genomic-data-standards/tximeta.png"></a> --- <img src="images/genomic-data-standards/linking-interfaces.svg" width="600" style="align:center"><br/> - Web standards and APIs (Data → Compute) --- ### The increasing importance of web standards As data and analysis become larger and more distributed, more recent standards increase involvement of web concepts --- ### Application programming interface (API) - An API defines protocols and formats for input and output of a computing system - It provides an abstraction layer to a user --- ### GA4GH:REFGET [Refget API Specification](http://samtools.github.io/hts-specs/refget.html) ```python GET /sequence/{id} # get sequence by ID. The primary method # for accessing specified sequence data. ``` --- ### GA4GH:DRS [Data Repository Service (DRS)](https://ga4gh.github.io/data-repository-service-schemas/preview/release/drs-1.0.0/docs/) > Provides a generic interface to data repositories so data consumers, including workflow systems, can access data in a single, standard way regardless of where it’s stored and how it’s managed ``` GET /objects/{object_id} # returns a JSON response including access methods ``` ``` /objects/{object_id}/access/{access_id} # returns a URL where you can get the object ``` --- ### GA4GH:DRS #### Current approach You share your data with a link on your website <div class="fragment"> #### Future approach You implement DRS. Then, a standard DRS client could get data, making your data more accessible. </div> --- ### GA4GH:WES [Workflow Execution Service](https://ga4gh.github.io/workflow-execution-service-schemas/docs/) Provides a standard way for users to submit workflow requests to workflow execution systems, and to monitor their execution - GET `/runs` - to list current runs - POST `/runs` - to start a new run - GET `/runs/{run_id}` - information about a run - GET `/runs/{run_id}/status` - status of a run - POST `/runs/{run_id}/cancel` - cancels a run --- ### GA4GH:TES [Task Execution Service](https://ga4gh.github.io/task-execution-schemas/docs/) - GET `/tasks` - list tasks tracked by the TES server - POST `/tasks` - create new task - GET `/tasks/{id}` - get a single task - POST `/tasks/{id}:cancel` - cancel a task --- ### Conclusion Who is the target audience? - humans? - computers? <div class="fragment">It's easy to build a human-friendly interface to data organized for computers. The opposite is not true.</div> <div class="fragment"> So, always build in this direction: computers → humans </div> --- 1. Make and share *everything* in *computable* form - your raw and processed data - your results - your writing (markdown) - computability precedes interoperability 2. Follow standard file formats 3. Use controlled terms (e.g. ontologies) 4. Identify elements with unique identifiers --- - Genomes will always be a community resource - It's a huge, worldwide, rapidly growing endeavor - Be familiar with existing standards - Use them whenever possible - Be aware of work on new standards <div class="fragment"> Remember, interoperability means impact </div>