## RNA-seq pseudoalignment, K-mers, and Hashing --- ## Lecture overview - RNA quantification with EM - RNA pseudoalignment - K-mer counting - Hashing and MinHash - Bloom filters --- ### Intro to RNA-seq <a href="https://en.wikipedia.org/wiki/RNA-Seq"><img src="https://upload.wikimedia.org/wikipedia/commons/f/f3/Summary_of_RNA-Seq.svg" height="600"></a> --- ### Transcript models <img src="images/genomic-intervals/brca2-gene-model.png" height="320"> RNA analysis may include: - de novo assembly - pre-existing fixed set for quantification --- ### Review of EM - Expectation Maximization is an algorithm to find Maximum Likelihood Estimates - It is used in situations where there is missing data - Missing data may be either intentional or unintentional - Intentional missing data is used for mixture models --- ### EM for mixture models $$ P(\mathbf{X}) = \prod_{i=1}^{N} \sum_{k=1}^{K} \pi_k P(X_i=x | \theta_k) $$ $P(\mathbf{X})$ is the joint probability of observed data $\mathbf{X}$ $\pi_k$ are the mixture parameters $\theta_k$ are the model parameters for each component <div class="fragment"> Latent indicator/responsibility variable $Z_{ik}$ is introduced to assign data $i$ to component $k$. </div> --- ### EM in RNA-seq - Observed data is a set of aligned reads $R$ - Latent mixture vars $Z$ assigns $R_j$ to transcript $T_i$ - We want to infer expression of each transcript $\theta_i$ - See: [Li 2010](https://doi.org/10.1093/bioinformatics/btp692) and [Li 2011](https://doi.org/10.1186/1471-2105-12-323) #### Expectation (E-step) Guess assigning reads to transcripts ($E[Z]$) #### M-step Calculate expression values ($\Theta$) for each transcript --- ### Expectation (E-step) Guess assigning reads to transcripts ($E[Z]$) #### E-step detail $E[Z] = P(Z=1|r,\theta_{i}^{old})$ - $Z_{ij}$ is the probability that read $j$ was drawn from transcript isoform $i$. - $Z$ is huge ($|R| \times |T|$), but we can use alignment to constrain it (fix some at $0$). --- #### E-step detail Just assign the read to the transcript according to its abundance estimate $\theta_i$ $E[Z] = P(Z=1|r,\theta_{i}^{old})$ $E[Z_{ij}] = \frac{\theta_i^{old}}{\sum_{i|Z_{ij}\not=0}{\theta_i^{old}}}$ - Uniquely aligning reads are assigned to 1 isoform, $E[Z_{ij}]=1$ - Multi-aligning reads are assigned proportionally to the expression estimates $\Theta$, for compatible transcripts. --- ### M-step Calculate expression values ($\Theta$) for each transcript $\argmax_{\Theta} \cal L(\theta_{i} | Z, r)$ #### M-step detail - We want to infer $\Theta = [\theta_1, \theta_2, ..., \theta_n]$ - $\theta_i$ measures expression for isoform $i$ $\theta_i^{new} = \frac{\sum_j^R{Z_{ij}}}{N}$ --- ### Summary of EM for RNA-seq E-step: Assigning reads to transcripts ($E[Z]$) $E[Z] = \frac{\theta_i^{old}}{\sum_{i}{\theta_i^{old}}}$ M-step: Calculate expression ($\Theta$) for each transcript $\theta_i^{new} = \frac{\sum_j^R{Z_{ij}}}{N}$ <div class="fragment"> Traditionally uses read-transcript alignments </div> --- # K-mers --- ### k-mers: subsequences of length *k* <div class="row"> <div class="col"> <h2>3-mers</h2>  </div> <div class="col"> <h2>2-mers</h2>  </div> </div> --- How many 7-mers are there in a sequence with $l=11$ bases? <div class="fragment"> $x = l-k+1 = 5$ ``` |ABCDEFGHIJK| |1-----7 | 1 | 1-----7 | 2 | 1-----7 | 3 | 1-----7 | 4 | 1-----7| 5 ``` </div> <div class="fragment"> This is total k-mers, not unique k-mers </div> --- ### Exponential growth $4^k$ ``` > cbind(k=seq(1,20), combinations=4^seq(1, 20)) k combinations [1,] 1 4 [2,] 2 16 [3,] 3 64 [4,] 4 256 [5,] 5 1024 [6,] 6 4096 [7,] 7 16384 [8,] 8 65536 [9,] 9 262144 [10,] 10 1048576 [11,] 11 4194304 [12,] 12 16777216 [13,] 13 67108864 [14,] 14 268435456 [15,] 15 1073741824 [16,] 16 4294967296 [17,] 17 17179869184 [18,] 18 68719476736 [19,] 19 274877906944 [20,] 20 1099511627776 ``` --- Human chromosome 1 has 248,956,422 bp. > Assuming a random distribution of 4 bases, what is the expected count of a given 7-mer on human chromosome 1? --- $l = 248,956,422$ size of chr1 $l-k+1 = 248,956,416$ total 7-mers in chr1 $4^7 = 16384$ possible 7-mer types $ 248,956,416 / 16384 \approx \mathbf{15195} $ --- ## Kmers and motifs <div class="row"> <div class="col"> <h2>K-mer</h2>  </div> <div class="col"> <h2>Motif</h2> <img src="images/k-mer-analysis/motif.png" height="200"> </div> </div> - A k-mer is like a simplified motif with no weights - K-mers are faster to compute and require less data - K-mers represent a single instance, rather than a set --- ## Applications of K-mers - Alignment seeds (fast RNA counting) - Counting for relationships (e.g. metagenomics) - Dinucleotide/codon analysis, background models - Sequence assembly (De Bruijn graphs) - Simplified transcription factor binding - RNA pseudoalignment --- ## K-mer Counting  <div class="mermaid" style="display:none"> graph TD GATTACAGATTCAG-->GAT; GATTACAGATTCAG-->ATT; GATTACAGATTCAG-->TTA; GATTACAGATTCAG-->TAC; GATTACAGATTCAG-->ACA; GATTACAGATTCAG-->ATT; GATTACAGATTCAG-->CAG; GATTACAGATTCAG-->AGA; GATTACAGATTCAG-->TTC; GATTACAGATTCAG-->TCA; GATTACAGATTCAG-->CAG; GAT-->n([1]); ATT-->n2([2]); TTA-->n3([1]); TAC-->n4([1]); ACA-->n5([1]); CAG-->n6([2]); AGA-->n7([1]); TTC-->n8([1]); TCA-->n9([1]); </div> K-mer counting converts a sequence into a vector  <div class="mermaid" style="display:none"> graph LR GATTACAGATTCAG-->n1["1 2 1 1 1 1 2 1 1 1"]; CTGAGGACTCGACT-->n2["2 1 0 0 2 2 3 2 2 1"];; </div> --- Remember: $4^k$ ``` > cbind(k=seq(1,20), combinations=4^seq(1, 20)) k combinations [8,] 8 65536 [9,] 9 262144 [10,] 10 1048576 [11,] 11 4194304 [12,] 12 16777216 [13,] 13 67108864 [14,] 14 268435456 [15,] 15 1073741824 [16,] 16 4294967296 [17,] 17 17179869184 [18,] 18 68719476736 [19,] 19 274877906944 [20,] 20 1099511627776 ``` High $k$ $\rightarrow$ high-dimensional vectors, sparsity --- ### Hashing > A hash function maps an object to a finite space - must be deterministic - should be very fast to compute - should reduce collisions Uses of hash functions 1. Hash table keys 2. Evaluating similarity (or identity) 3. Cryptography --- 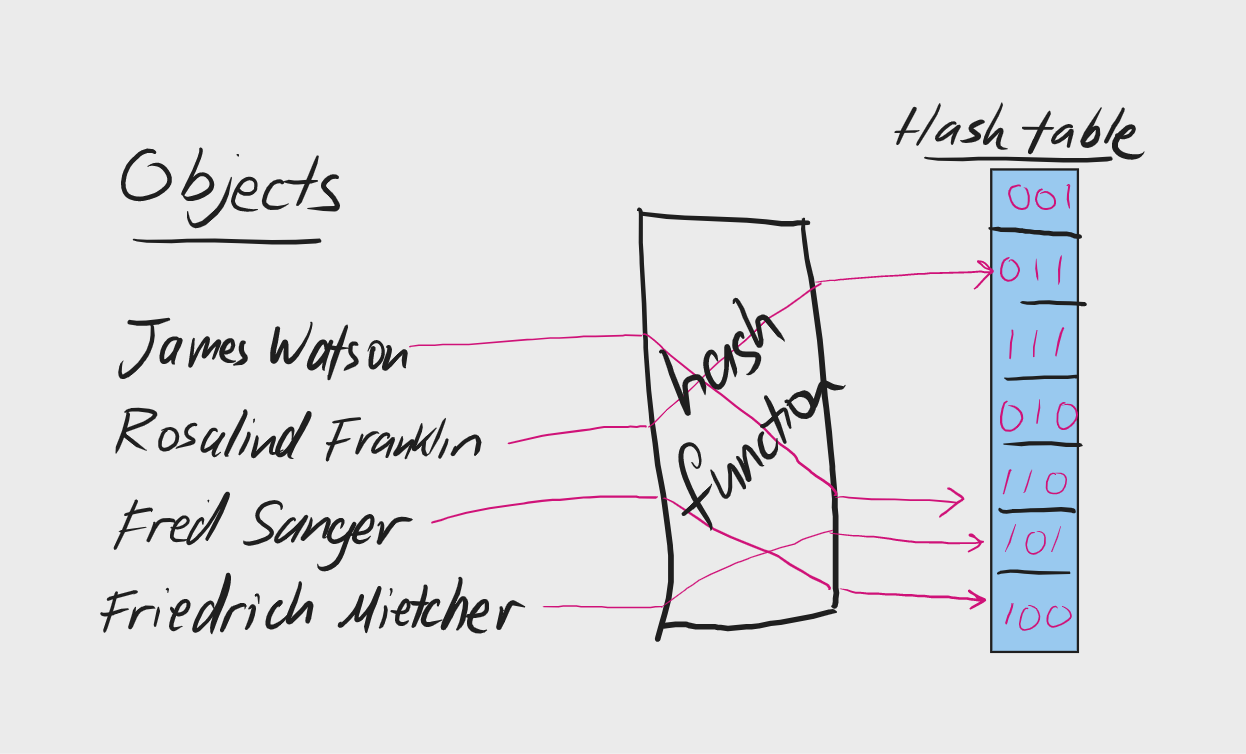 --- ### Hash tables - provide $O(1)$ complexity for item access - Can store large objects for lookup with a key --- ### Cryptographic hash function - a subset of hash functions with particular purposes: - designed to go one way (hard to de-hash) - designed to avoid collisions - relies on the avalanche effect (small change in inputs leads to large change in output) --- ### md5 algorithm- "Message Digest" In 2011, the Internet Engineering Task Force (IETF) [advised in this document](https://tools.ietf.org/search/rfc6151) that > The published attacks against MD5 show that it is not prudent to use MD5 when collision resistance is required. --- ## MinHash MinHash uses hashing to determine similarity between two sets very quickly. <div class="fragment"> Jaccard index measures similarity between two sets $$ J(A,B) = \frac{|A \cap B|}{|A \cup B|} $$ Computing can be time-consuming for large sets </div> <div class="fragment"> MinHash estimates the Jaccard index rapidly, using hashing to avoid computing intersection and union </div> --- ### MinHash $U$ is the universe of elements Hash function $h$ maps set elements to unique integers $h(x): U \rightarrow \mathbb{Z}$ Given a subset $S \subset U$, $h_{min}(S)$ is the minimum hash value (integer) $h(e_i)$ for each element $e_i \in S$. --- $h_{min}(A) = h_{min}(B)$ if and only if the element with minimum hash value in $U = A \cup B$ is also in $A \cap B$. <div class="fragment"> The probability of this is $J(A,B) = \frac{|A \cap B|}{|A \cup B|}$. So $P(h_{min}(A) = h_{min}(B))$ is an estimator of $J$. </div> <div class="fragment"> But we need more than one draw: - multiple hash functions - take the $n$ lowest as the MinHash *signature* or *sketch* </div> --- MinHash has been used for fast comparisons of DNA sequences with k-mers. - Metagenomics ([Ondov 2016](https://doi.org/10.1186/s13059-016-0997-x)) - Sourmash ([Brown 2016](https://doi.org/10.21105/joss.00027)) - Phylogenetics ([Moi 2020](https://doi.org/10.1371/journal.pcbi.1007553)) - Genome assembly ([Berlin 2015](https://doi.org/10.1038/nbt.3238) and [Koren 2017](https://doi.org/10.1101/gr.215087.116 )) --- ## Bloom filters - Probabilistic membership testers using hash tables - Fast and memory efficient - Guarantee an item *is not* in the set, but not if it is (false positives allowed) - Elements cannot be removed from the set --- ### Bloom filter components 1. A bit vector $F$ of length $l$ (size of the filter) 2. $k$ hash functions $h_i(x): U \rightarrow \mathbb{Z}$, $i \in 1, 2, ..., k$, which map objects in the universe $U$ to integers $\mathbb{Z}$ in $(0, l]$. Parameters: - Size $l$: how many elements in the bit vector? - Number of hash functions - Hash functions themselves --- ### Bloom filter procedure: add A Bloom filter represents a set of elements $S \subset U$ To add an element $e$: 1. Compute the $k$ hash functions, $h_i(e), ...$ 2. Set $F[h_i(e)] = 1$ for $i \in 1:k$. --- ### Bloom filter procedure: test To test for presence of an element $e$: 1. Compute the $k$ hash functions, $h_i(e), ...$ 2. Test $F[h_i(e)] \overset{?}{=} 1$ for $i \in 1:k$. 3. if $\forall i \in [1, k]: F_i=1$, then $e \overset{?}{\in} S$. Otherwise, $e \not\in S$. ---  --- ## A basic hash function $h(x): U \rightarrow \mathbb{Z}$, $\mathbb{Z} \in (0, l]$ ```R hashfunc = function(d) { hash = 0 r = as.integer(charToRaw(d)) # convert char to int for (item in r) { hash = hash * 7 + item } hash %% 1024 } ``` --- ## Hash function considerations - uniformity of hash values - more functions affects speed, accuracy, and fill rate <div class="fragment"> [Kirsch-Mitzenmacher optimization](https://www.eecs.harvard.edu/%7Emichaelm/postscripts/tr-02-05.pdf) Given 2 hash functions $h_1(x)$ and $h_2(x)$, simulate additional values with $g_i(x) = h_1(x) + i * h_2(x)$, $i \in [0, k-1]$ for $k$ additional hash functions. </div> --- ### Bloom filters in practice - RNA-seq quantification ([Zhang 2014](https://doi.org/10.1093/bioinformatics/btu288)) - Pre-alignment species filtering ([Chu 2014](https://doi.org/10.1093/bioinformatics/btu558)) - Single-cell transcriptome assembly ([Nip 2020](https://doi.org/10.1093/10.1101/gr.260174.119)) - Large-scale transcript searching ([Solomon 2016](https://doi.org/10.1038/nbt.3442)) - Genome assembly ([Jackman 2017](10.1101/gr.214346.116 )) - Review of [Sketching and Sublinear Data Structures in Genomics](https://www.annualreviews.org/doi/abs/10.1146/annurev-biodatasci-072018-021156) --- ## RNA Pseudoalignment Quantifying expression... - only requires an assignment of reads to transcripts - therefore doesn't require a complete alignment - could be sped up by assigning reads without computing an alignment --- ## RNA Pseudoalignment Build index: 1. de Bruijn graph 2. contigs 3. k-mer index Pseudo-align reads: 1. Contig lookup 2. Intersection --- Transcriptome de Bruijn graph (T-DBG) 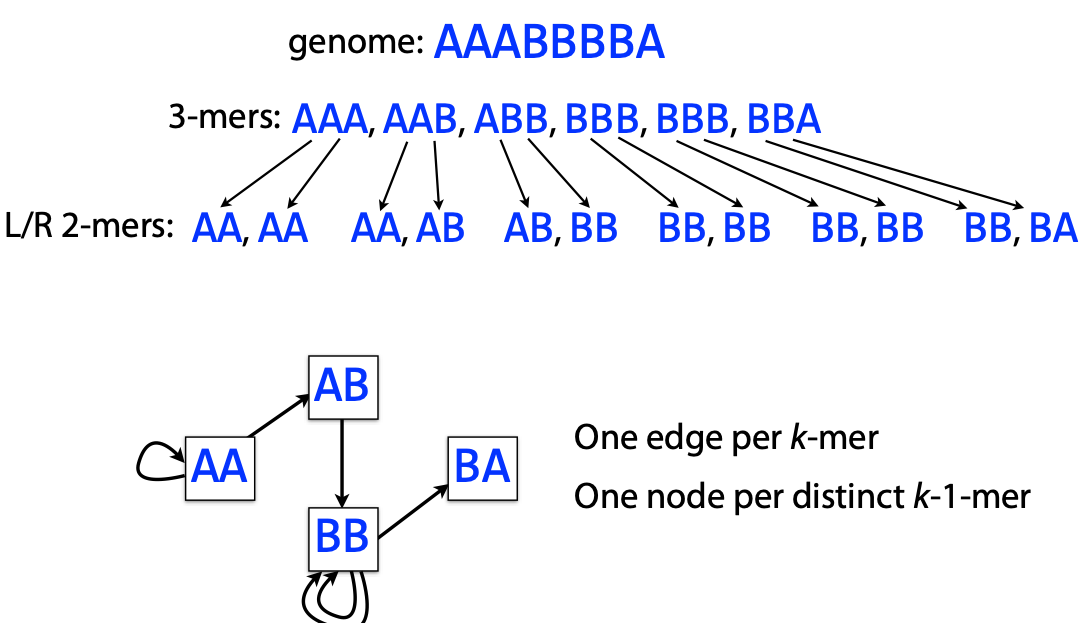 --- ### Kallisto <a href="https://www.nature.com/articles/nbt.3519/figures/1"><img src="images/k-mer-analysis/kallisto_fig1a.png" width="500"></a> - each node corresponds to a k-mer (default k=31) - isoforms are overlaid on the graph --- <a href="https://www.nature.com/articles/nbt.3519/figures/1"><img src="images/k-mer-analysis/kallisto_fig1c.png" width="400"></a> - linear stretches with the same isoforms are 'contigs' - Build Index: A hash table maps the K-mer to contigs <div class="fragment"> Align: 1. Look up compatibility class for each k-mer in read 2. Intersect them. </div> --- ### Quantify $$ \cal L(\alpha) \propto \prod_{r \in R} \sum_{t \in T} y_{rt} \frac{\alpha_t}{l_t} $$ reads $r \in R$, transcripts $t \in T$, compatibility matrix $y_{rt}$, expression levels $\alpha_t$ for transcript $t$ with length $l$ --- ## Pseudoalignment summary Assign reads to transcript with fast k-mer hash tables. Use EM to iteratively compute assignments $Z$ (E-step) Then maximize by computing expression levels $\Theta$